Structural and Functional Studies of Proteins Involved in Bacterial Conjugation
Bacterial conjugation is the process by which DNA can be transferred from one bacterium (donor) to another (recipient). This process enables the distribution of genetic material throughout a bacterial population, and is a mechanism for bacterial evolution and adaptation to unique environments. It is also a mechanisms by which genes encoding antibiotic resistance and other virulence factors are spread through a bacterial population. Related systems are also used by several pathogens to secrete virulence factors into the medium (for instance the toxin from B. pertussis, responsible for whooping cough) or to directly inject them into host cells (for instance CagA from the human pathogen H. pylori or the T-DNA of the plant pathogen A. tumefaciens). These systems have been classified a subgroup of the type IV secretion system (T4SS) based upon the similarities between the genes involved in the processes.
The F-plasmid of E. coli, first described in in the 1940s, is the prototypical system for bacterial conjugation. However, despite a large accumulation of genetic and biochemical data over the last half century, a detailed structural analysis of the component proteins of this system remains elusive, especially for proteins involved in regulating the conjugation process (there are some very nice studies of T4SS in membranes etc.). Our research is focused on examining proteins that make up the F-plasmid T4SS from a structural and functional standpoint, with an aim to understand how the T4SS component proteins interact to form a complex molecular pump to transfer DNA from the donor to the recipient cell.
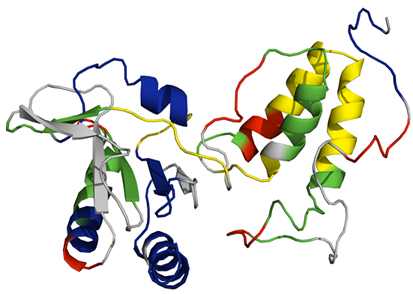
One protein we have been exploring is TraF, which is a hallmark protein of F-like T4SSs. While the crystallographic structure of the protein remains elusive, we have recently reported the a dynamic analysis of the protein using Time Resolved Hydrogen Deuterium Exchange Mass Spectrometry (HDX-MS). Shown above is a dynamic mapping of TraF based on a predicted model of the protein (Lento et al. FEBS Letters 590(3), 376-386 (2016)). It was observed that the C-terminal domain of the protein (on the left of the image above), which is a thioredoxin-like domain without the active CxxC motif, is more stable that the N-terminal domain (on the left) that is predicted to make interactions with TraH for localization of TraF to the outer membrane.